Expert-supervised-AI Cycle from Hypothesis to Real-World Materials Discovery
A vision of AI in materials science promises discoveries on an unprecedented scale. With algorithms now capable of generating thousands of candidate compositions, breakthroughs in superconductors, next-generation batteries, or water-splitting catalysts feel closer than ever. Yet a growing danger lies in science being drawn into a maze of digital possibilities — predictions that dazzle on screen but risk leading to nominal advances and neglected experimental insight. To ensure genuine progress, computational proposals must operate within a cycle of hypothesis, expert scrutiny, and experimental validation, balancing abstract predictions with the realities of chemistry, stability, and application. By treating AI’s output as provisional guidance rather than prescribed outcome, we can establish a human–AI discovery workflow that leverages digital tools1–4 to navigate the complexity of materials space (Figure 1).
In inorganic materials science, decades of mineralogical and synthetic research have yielded more than 200,000 experimentally verified structures5. This collection forms the foundation of most AI applications in the field — from exploring elemental substitutions to predicting functional properties6. Yet, extensive as it is, this dataset constrains modeling efforts and can bias algorithms toward familiar patterns. This reemphasizes the importance of expert judgment and experimental validation to ensure that AI-assisted discovery extends knowledge rather than merely reproduces it. Elemental substitution within known compositions and structures is a long-established design strategy and a convenient starting point for AI modeling7. However, this “design by analogy” depends on prior discoveries of parent materials. Its evolutionary nature often leads to incremental modifications rather than truly novel structures or properties8,9.
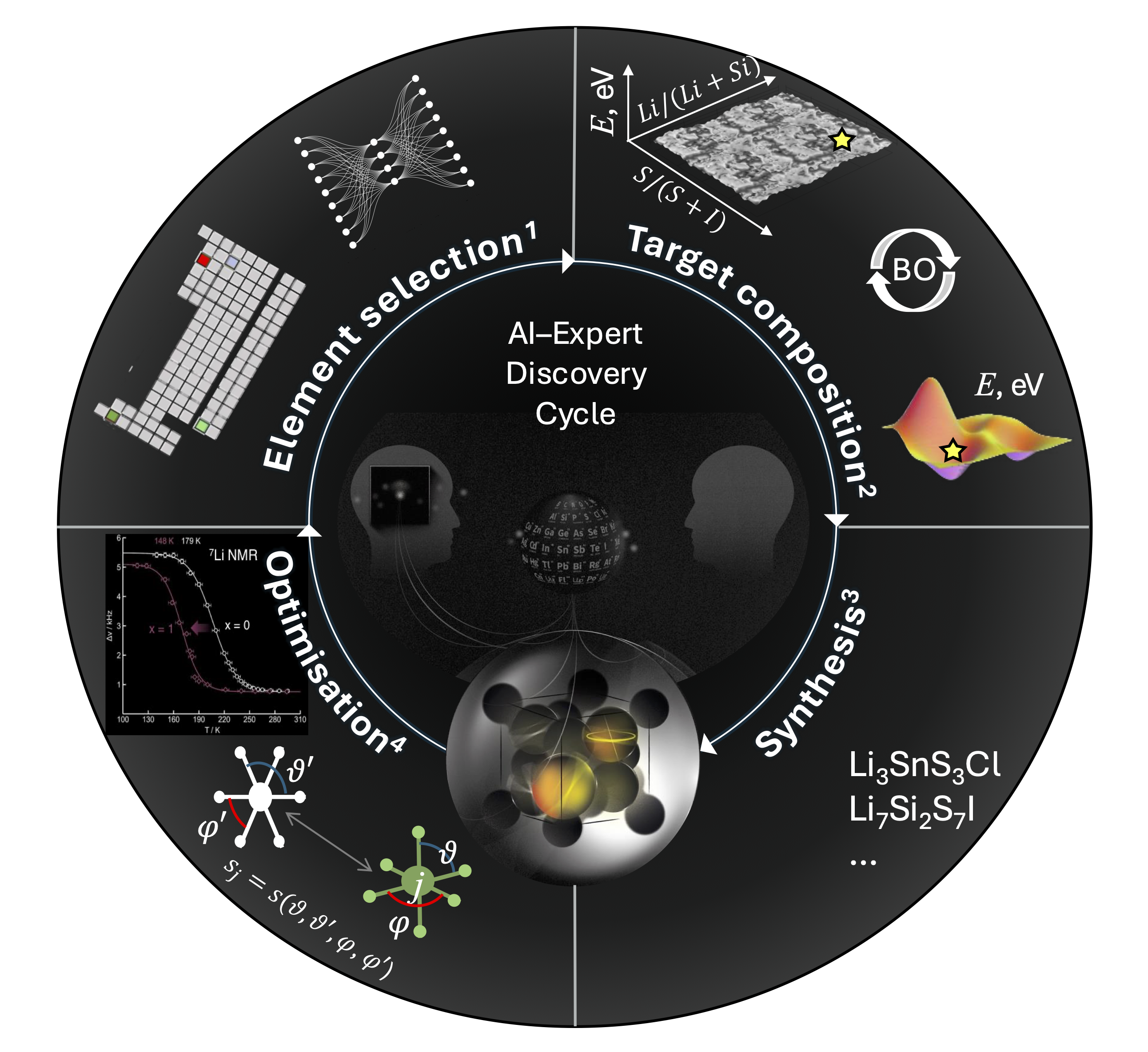
.
To explore beyond this foundational dataset, we developed tools to evaluate the synthetic accessibility and chemical novelty of previously uncharted combinations of elements — ultimately addressing a core question of chemistry: “How to choose?”1. Leveraging a variational autoencoder (VAE) for unsupervised pattern recognition in a high-dimensional chemical space, we rank hypothetical elemental combinations by similarity to known compounds, thereby guiding attention toward promising yet unexplored regions VAE-selected systems can then be prioritized for functionality through explainable AI (XAI), which reveals links between element choice and anticipated material properties10. With domain expertise, this process can be directed toward practical targets — for example, combinations of lithium, one frame-forming metal, and two anions with geometries favorable for ionic mobility. Such reasoning led to the hypothesis and subsequent experimental discovery of a novel superionic conductor, Li7Si2S7I (LSSI)3.
Exploration of compositional space via crystal structure prediction (CSP) typically relies on quantum-mechanical calculations and unit-cell approximations, which constrain searches to stoichiometries expressible as small integer ratios of atoms. To access the traditionally “incomputable” regions between these points, we hypothesized a continuous energy–composition relationship and applied AI-guided (Bayesian optimization) to target structures offering the greatest informational gain2. The resulting energy–composition maps identify near-stable compositions at a fraction of the computational cost, focusing experimental efforts on the most promising chemical regions. At this stage, expert supervision closes the discovery loop: domain knowledge filters AI suggestions through mechanistic reasoning — for instance, prioritizing systems with anion-size asymmetry as a design rule for superionic conduction. Guided by this insight, we selected the S–I system from among several VAE-ranked candidates (Li–Si–S–Cl, Li–Si–S–Br, Li–Si–S–I), leading to the synthesis of LSSI3.
Newly discovered materials can serve as prototypes for subsequent optimization. In LSSI’s case, substitution or doping strategies can be directed by statistical and AI-based analyses of local geometric motifs11. This optimization stage produced Li7Si2–xGexS7I, where partial Ge substitution enhances Li conductivity at lower temperatures — an outcome of both AI guidance and expert experimental intuition4.
Materials discovery increasingly benefits from the synergy between human expertise and AI tools, which together expand our capacity to tackle the complexity and scale of materials science. Emerging methods - including generative models, large language models, and high-throughput simulations — offer exciting opportunities for scientific acceleration that align with societal and industrial needs. Yet, dedicating vast resources to these tools alone does not ensure progress; without careful guidance, such approaches risk prioritizing numerical patterns over genuine scientific understanding. This highlights the enduring importance of expert supervision and the development of explainable AI to interpret predictions and reveal underlying principles. As XAI matures and fosters transparent hypothesis exchange within multi-agent discovery loops, materials research can evolve into a truly collaborative process — one that reshapes both scientific understanding and real-world innovation.
References
- Vasylenko A., Gamon J., et al. Nat. Commun. 2021; 12(1):5561.
- Vasylenko A., Asher B.M., et al. J. Chem. Phys. 2024; 160(5):054110.
- Han G., Vasylenko A., Daniels L.M., et al. Science 2024; 383(6684):739.
- Han G., Daniels L.M., Vasylenko A., et al. Angew. Chem. Int. Ed. 2024; 67(37):e202409372.
- Zagorac D., Müller H., et al. J. Appl. Cryst. 2019; 52(5):918.
- Agrawal A., Choudhary A. APL Mater. 2016; 4(5):053208.
- Merchant A., Batzner S., et al. Nature 2023; 624(7990):80.
- Cheetham A.K., Seshadri R. Chem. Mater. 2024; 36(8):3490.
- Antypov D., Vasylenko A., et. al. Acc. Chem. Res. 2025; 58(9):1355.
- Vasylenko A., Antypov D., et al. NPJ Comput. Mater. 2023; 9(1):1.
- Vasylenko A., Antypov D., et al. Digit. Discov. 2025; 4(2):477.